|
|
|

ELA - Una enfermedad con mucha esperanza
|
¿Qué es la esclerosis lateral amiotrófica? Se buscará informar acerca de una enfermedad que se encontró en boca de muchos a partir de la práctica impulsada en Boston, Estados Unidos por un ex jugador de béisbol, College Pete Frates, (una de las víctimas de la enfermedad) llamada ?Ice Bucket Challenge?, donde se arrojan un recipiente de agua helada sobre la cabeza y se reta a tres nuevas personas a cumplir el desafío donando 10 dólares o, en caso de no realizar el ?baño?, donar 100 dólares a alguna institución que investigue acerca de esta patología. La campaña creció tanto que muchas reconocidas personalidades como, por ejemplo, el hoy en día más mencionado jugador de futbol a nivel mundial, Lionel Messi, el presidente de los Estados Unidos Donald Trump o la afamada cantante Shakira formaron parte.
Agregado: 14 de DICIEMBRE de 2021 (Por MarÃa Victoria Guntren) | Palabras: 10628 | Votar | Sin Votos | Sin comentarios | Agregar Comentario
Categoría: Apuntes y Monografías > Medicina >
Material educativo de Alipso relacionado con ELA Una enfermedad con mucha esperanza
Enlaces externos relacionados con ELA Una enfermedad con mucha esperanza
Autor: María Victoria Guntren (vico.mguntren@gmail.com)
 Este apunte fue enviado por su autor en formato DOCX.
Para poder visualizarlo correctamente (con imágenes, tablas, etc) haga click aquí o aquí si desea abrirla en ventana nueva. Este apunte fue enviado por su autor en formato DOCX.
Para poder visualizarlo correctamente (con imágenes, tablas, etc) haga click aquí o aquí si desea abrirla en ventana nueva. |
<
Esclerosis Lateral Amiotrófica
Una enfermedad con mucha esperanza

Guntren, María Victoria
Noviembre 2017
Instituto de Formación Técnica Superior.
Buenos Aires, Argentina
Epidemiología.
Un trastorno muy conocido a partir de la campaña “Ice Bucket Challenge” pero a la vez son muy pocos los que saben que esta enfermedad existe. Considerada rara, es una de las más crueles patologías, que perturba sin hacer distinción de etnias, género o edad.
“Espero mi ejemplo sirva de estímulo y de esperanza para otros en una situación similar ¡Nunca se rindan!” -Stephen Hawking, 2012-
Contenido
Esclerosis lateral amiotrófica y Esclerosis Múltiple: Diferencias. 8
¿Qué Es La Esclerosis Lateral Amiotrófica?. 9
Tipos de Esclerosis Lateral Amiotrófica. 14
Steven Hawking: Ejemplo de lucha y esperanza. 58
¿Cómo inscribirse al ReNELA?. 67
Resumen
La esclerosis lateral amiotrófica, conocida como enfermedad de Lou Gehrig (haciendo referencia a un afamado jugador de béisbol afectado por la enfermedad) o enfermedad de Charcot (en honor al primer médico francés que documento acerca de esta patología) es un trastorno neurodegenerativo progresivo que suele ser mortal, provocado por una degeneración de las motoneuronas que produce debilidad muscular, atrofia y rigidez.
Aún no se conocen las causas que generan esta patología, sino que se cree que es una mezcla de factores, especialistas presumen que afecta tres veces más a hombres que a mujeres y aseguran que no es una enfermedad contagiosa.
Sus síntomas varían de un paciente a otro, algunos tienen un curso más rápido y lo que el paciente comienza a notar es que sus movimientos voluntarios disminuyen conforme va pasando el tiempo, respetándose los movimientos oculares, los órganos de los sentidos y los esfínteres.
Actualmente el diagnóstico es puramente clínico, sólo los idóneos en neurología lo establecen y no hay prueba de laboratorio que pueda confirmar el diagnóstico de esclerosis lateral amiotrófica.
En el día de la fecha no hay un tratamiento curativo efectivo contra la ELA, pero si medicamentos que logran ralentizar el proceso de degeneración de las neuronas afectadas.
Abstract
Amyotrophic lateral sclerosis, known as Lou Gehrig's disease (referring to a famous baseball player affected by the disease) or Charcot's disease (in honor of the first French doctor who wrote about this pathology) is a progressive neurodegenerative disorder that usually be mortal, caused by a degeneration of motor neurons that produces muscle weakness, atrophy and stiffness.
The causes of this pathology are still unknown, but it is believed that it is a mixture of factors, specialists presume that it affects three times more men than women and assure that it is not a contagious disease. Its symptoms vary from one patient to another, some have a faster course and what the patient begins to notice is that their voluntary movements decrease as time passes, respecting eye movements, sense organs and sphincters.
Currently the diagnosis is purely clinical, only those qualified in neurology establish it and there is no laboratory test that can confirm the diagnosis of amyotrophic lateral sclerosis.
In the day of the date there is no effective curative treatment against ALS, but drugs that manage to slow the process of degeneration of affected neurons.
Objetivo
¿Qué es la esclerosis lateral amiotrófica? Se buscará informar acerca de una enfermedad que se encontró en boca de muchos a partir de la práctica impulsada en Boston, Estados Unidos por un ex jugador de béisbol, College Pete Frates, (una de las víctimas de la enfermedad) llamada “Ice Bucket Challenge”, donde se arrojan un recipiente de agua helada sobre la cabeza y se reta a tres nuevas personas a cumplir el desafío donando 10 dólares o, en caso de no realizar el “baño”, donar 100 dólares a alguna institución que investigue acerca de esta patología. La campaña creció tanto que muchas reconocidas personalidades como, por ejemplo, el hoy en día más mencionado jugador de futbol a nivel mundial, Lionel Messi, el presidente de los Estados Unidos Donald Trump o la afamada cantante Shakira formaron parte.
¿Hay diferentes tipos? ¿Cuáles son los síntomas que identifican a la ELA? ¿Existen métodos preventivos o procedimientos curativos? Se procurará comunicar en detalle las características que implica padecer ELA, cómo son las señales que presenta y cuáles son los cuidados que necesita un paciente con esta enfermedad, mencionar los tipos de alimentación que ayuda a llevar adelante el proceso de la afección, con que medicamentos y/o tratamientos podemos ofrecer mejor calidad de vida desacelerando el progreso de este tipo de esclerosis y su sintomatología.
Marco Teórico I
Esclerosis lateral amiotrófica y Esclerosis Múltiple: Diferencias
Actualmente se confunden ambas enfermedades dado que ambas se llaman “Esclerosis” y son enfermedades neurodegenerativas, pero ciertamente cuentan con muchas grandes diferencias las cuales se destacan a continuación para poder comprender sus conceptos.
La Esclerosis Múltiple es una enfermedad neurodegenerativa, crónica, con origen desconocido que afectan al sistema nervioso central, dichas características son muy similares a la Esclerosis Lateral Amiotrófica.
La diferencia fundamental con ELA es que la EM es una enfermedad autoinmune, es decir que el sistema inmunológico quien normalmente nos defiende de agentes extraños daña una parte de nuestro organismo, en este caso daña la mielina que es la capa que protege las fibras nerviosas del Sistema Nervioso Central (SNC) – cerebro, medula espinal y nervios ópticos- sin causa conocida hasta el día de hoy, provocando que la habilidad de los nervios de conducir impulsos eléctricos desde y hacia el cerebro se interrumpa. Esta desmielinización y cicatrización se presentan en distintos momentos y en zonas cambiantes por eso recibe el nombre: esclerosis múltiple.
EM no es una enfermedad contagiosa, por el momento no tiene cura, sino que sólo cuenta con medicamentos atenuantes que enlentecen el progreso de la patología o disminuyen la frecuencia de brotes y no es mortal.
¿Qué Es La Esclerosis Lateral Amiotrófica?
Enfermedad de la Neurona Motora (ENM)- nombre con el cual se la conoce en Australia e Inglaterra- también conocida en Francia como enfermedad de Charcot, en honor al médico neurólogo nacionalizado allí que fue el primero que escribió sobre esta patología en 1869, o conocida en Norteamérica como enfermedad de Lou Gehrig haciendo homenaje al popular beisbolista estadounidense a quien en la famosa Clínica Mayo le fue confirmada el 19 de junio de 1939 e informándole una expectativa de vida de tres años que no llegó a cumplir dado que falleció el 2 de junio de 1941; son algunos de los nombres que recibe esta enfermedad que se caracteriza por el deterioro de las neuronas motoras superior e inferior, y lleva a la muerte al 50% de los enfermos dentro de los primeros 3 a 5 años luego del diagnóstico.
Para poder comprender la afección que genera la esclerosis lateral amiotrófica en nuestro cuerpo es necesario saber cómo funciona nuestro sistema nervioso, uno de los sistemas más importantes para nuestra vida. Este sistema, mediante las células especializadas para transmitir señales nerviosas -las neuronas-, procesa coordina e integra señales que nos llegan desde el medio externo, a través de nuestros órganos de los sentidos.
Este mecanismo comienza en el encéfalo, que forma parte del sistema nervioso central y está dividido en el cerebro, el cerebelo y el tronco del encéfalo, conectado con la médula espinal.
Aquí el accionar nervioso varía según el
tipo de movimiento a realizar ya que no es lo mismo el movimiento de caminar
(voluntario) como el de respirar o el del latido del corazón (involuntarios).
Al realizar un movimiento voluntario la primer motoneurona actuante es aquella
proveniente del encéfalo, a la que se denomina motoneurona superior, que será
la que con su axón recorre diferentes estructuras hasta llegar a la medula
espinal donde se contacta con la segunda motoneurona denominada “motoneurona
inferior” (este contacto entre neuronas se denomina sinapsis) y esta última
actúa sobre las fibras musculares a través de la acción electro-química
liberando acetilcolina y en cadena de ésta se libera calcio provocando que dos
proteínas presentes – actina y miosina - se contraigan acortando en simultaneo
todas las células presentes en el musculo, lo cual genera el movimiento.
La esclerosis lateral amiotrófica afecta de forma selectiva a las motoneuronas
tanto superior como inferior, provocando rápidamente disminución del movimiento
hasta la rigidez de los músculos de manera irreversible.
El nombre que define a la enfermedad proviene de las siguientes palabras griegas:
· "a" de "sin"
· "mio" de músculo
· "trófica" de nutrición
· "lateral" de lado (de la médula espinal)
· "esclerosis" de endurecimiento o cicatrización
o Amiotrófica: significa que los músculos han perdido la capacidad de nutrirse.
o Lateral: significa que la enfermedad afecta a los lados de la médula espinal, donde se encuentran los nervios que alimentan a los músculos.
o Esclerosis: significa que en la parte de la médula espinal afectada por la enfermedad se desarrolla un tejido endurecido o cicatrizal en vez de nervios sanos.
¿Por qué Se Produce ELA?
Actualmente el origen de esta enfermedad se encuentra en estudio, existen diversas hipótesis que intentan explicar el causante, pero aún no hay datos precisos. Se acentúan en estas hipótesis factores genéticos, ambientales (como infecciones virales y alteraciones minerales), alteraciones en el metabolismo, entre otros, aunque la combinación de diversos factores es la afirmación que demuestra tener mayor significación.
Factores ambientales
Surge la necesidad de realizar un estudio que describa el comportamiento de la enfermedad en el país. Los estudios existentes en la literatura mundial son de utilidad como parámetro orientativo, pero no podemos afirmar ni descartar que el comportamiento de la ELA sea el mismo en Argentina o en las diferentes regiones que la componen.
Hay un estudio de especial relevancia: el caso de la ELA endémica del Pacífico Occidental. En 1898 la Isla de Guam ubicada en el archipiélago de las Marianas, dejó de formar parte de la corona española y pasó a depender de Estados Unidos, mientras que el resto de las islas fueron vendidas a Alemania hasta después de la Segunda Guerra Mundial que comenzó a ser un estado asociado a Estados Unidos. Durante esa guerra Guam fue poblada por japoneses, pero a principios del siglo XX, estaba formada por “Chamorros” (nativos), españoles y pobladores iberoamericanos, mexicanos y procedentes de Filipinas.
ELA en esta isla es conocida como “lítico”, y denota su primer paciente en 1904, enunciando su epidemia en la década de 1940. En Guam se planteó inicialmente que la posibilidad de ELA fuera de origen tóxico producido por la ingesta de cicad (una semilla que contenía neurotoxinas como la beta-metil-aminoalanina (BMAA) o la cicasina y su derivado metilazoximetanol (MAM)) hecha harina ingerido por los nativos mediante tortillas, y existe una convicción de que a medida que fueron adaptando distintos hábitos occidentales comenzó a disminuir la presencia de esta patología. Este hecho ha forzado la hipótesis de que el origen de la enfermedad se encuentra especialmente vinculado con la toxicidad local adquirida a través de la alimentación.
Otro claro ejemplo de esta sospecha deviene a partir de la ingesta de un manjar exquisito para los Chamorros, los zorros voladores (un animal vegetariano que se alimenta de cicad), y que acumula y multiplica los neurotóxicos en su grasa) a lo cual se le sumaba la difusión de armas de fuego que supuso el aumento de caza hasta prácticamente la extinción de los mismos.
ELA en nuestro país, al igual que otras enfermedades neurodegenerativas, es una afección sin notificación obligatoria a nivel nacional porque no es una enfermedad transmisible, siendo estas últimas a lo que están dedicados los sistemas de vigilancia de enfermedades. Esto es el problema principal que nos imposibilita conocer cómo se distribuye o cómo se comporta a nivel poblacional y poder obtener datos que nos orienten de su causalidad.
Factores Genéticos
Comprende el 10% de los pacientes que padecen esta enfermedad. Se vincula a una herencia autosómica, es decir, que no está enlazada al sexo, y dominante por lo cual requiere la mutación de un único gen. Esto ocurre cuando el paciente cuenta con un historial de antecedentes familiares, es decir, que si tanto abuelo como padre desarrollaron la enfermedad, puede dar lugar a que su descendencia también la padezca.
La teoría que cobra más fuerza hace referencia al gen SOD1 descubierto 1993, situado en el cromosoma 21 que se encarga de codificar una la enzima “súper óxido dismutasa Cu-Zn” para controlar el metabolismo del oxígeno.
Alteraciones al metabolismo
Glutamato
El glutamato es un aminoácido esencial necesario para la transmisión de impulsos nerviosos, es el excitador más habitual del cerebro. Esta hipótesis define que el exceso de glutamato provocado por un defecto en su transporte origina la entrada masiva de calcio en la célula y esto lleva a la apoptosis (muerte neuronal).
Hipótesis estrés oxidativo y radicales libres
El mecanismo mejor descrito del papel que juega el estrés oxidativo es el que hace referencia a las mutaciones del gen SOD1, específicamente a una acumulación de los radicales libres, producto de la disfunción de esta enzima, lo cual provoca a su vez la muerte celular.
Otros autores mencionan que existen defensas antioxidantes inadecuadas que influyen tanto en la ELA familiar como esporádica a pasar de la falta de defecto en el gen SOD1 y a partir de allí se realizaron investigaciones clínicas y terapias en la enfermedad, pero no se ha logrado evidencia sobre su efectividad.
Alteración del metabolito de los neurofilamentos
Los neurofilamentos de las neuronas motoras se encuentran en la cola de la célula, es decir, si ELA provoca una alteración en las proteínas responsables del mantenimiento de la estructura de la neurona se imposibilita la transmisión.
Defectos en las mitocondrias
En ensayos clínicos realizados en ratones con mutaciones del gen SOD1 se han observado algunos marcadores de la degeneración mitocondrial que se han exacerbados en el inicio de la enfermedad. Esta disfunción se acompaña del incremento en la producción de radicales libres, lo cual se relaciona con la teoría antes descripta.
Hay otras hipótesis relacionadas a la etiología de la enfermedad de la motoneurona, sin embargo, aún están en proceso y no dan lugar a que se puedan demostrar satisfactoriamente en pacientes humanos.
Tipos de Esclerosis Lateral Amiotrófica.
La clasificación clínica se realiza a partir de si se trata de ELA heredada o adquirida y por la ubicación de la motoneurona afectada (inferior o superior) al momento del inicio de la enfermedad. De esta manera se la difiere en:
Esclerosis Lateral Amiotrófica Esporádica
Hace referencia a aquella ELA adquirida, que alude al 90% de las personas con esta enfermedad, caracterizada por aparecer mayormente entre los 30 y 60 años con mayor cantidad de casos en hombres que mujeres y, sobre todo, en personas que practican deportes de mayor contacto, como artes marciales mixtas o boxeo, y traumatismos en la medula espinal, o personas con familiares ascendentes directos con trastornos mentales. La causa en esta tipología es desconocida; sin embargo, hasta un 7% también puede deberse a la expansión de hexa-nucleótidos en individuos caucásicos de origen Europeo del Norte. Dentro de ésta tenemos un suborden:
Clásica: En este tipo de ELA se presentan síntomas combinados de la alteración en la neurona superior e inferior, lo cual les genera debilidad y deterioro de las extremidades, rigidez muscular o calambres. Algunos pueden notar que tropiezan cuando caminan o que se les caen las cosas.
Con parálisis
bulbar progresiva
Involucra
un daño tanto en las neuronas superiores como inferiores. La afección comienza
en los músculos conectados a través de los nervios craneales controlan la
articulación, masticación, deglución, y luego afecta a los demás músculos
debilitando el organismo de forma generalizada.
Con atrofia
muscular progresiva
Comienza causando daño principalmente en la motoneurona inferior. Se ve
reflejada en la torpeza con las manos, debilidad o reflejos disminuidos.
Con esclerosis
lateral primaria
Comienza con síntomas de la motoneurona superior, constituyendo el tipo de ELA
más raro de todos. Causa debilidad en extremidades inferiores así como puede
también reflejarse en torpeza en las manos o el habla, y los reflejos se tornan
exagerados.
Esclerosis
Familiar (hereditaria)
Se
transmite con herencia autosómica dominante por un gen alterado en el brazo
largo del cromosoma 21
Siendo esta última causada en un 20% por mutaciones en el gen de la superóxido dismutasa 1 (SOD1). Adicionalmente, evidencias de familias con miembros afectados de ELA, otros afectados con Demencia Frontotemporal (DFT) y otros con DFT asociada a signos de motoneurona, confirman la existencia de un espectro entre la ELA y la DFT. El análisis de ligamiento de estas familias identificó un locus en el brazo corto del cromosoma 9 que recientemente fue descubierto como una expansión de hexanucleótidos no codificantes GGGGCC en el gen C9ORF726. Datos recientes adjudican hasta un 40% de las formas familiares de ELA o DFT a dicha expansión.
ELA en “Racimos”
También conocida
como “territorial” o “Guameña”, se debe a que se ha presentado un número de
casos extraordinariamente altos, distinguiéndose en esta clasificación algunos
brotes como los del Pacífico suroeste: Isla de GUAM, Península de Kii de Japón
y algunos grupos del oeste de Nueva Guinea.
Marco Teórico II
Los primeros síntomas
La enfermedad comienza a exponerse de una u otra forma de acuerdo con el tipo de ELA, antes mencionados, que se desarrolle:
Síntomas a partir de la afección de la motoneurona superior.
Espasticidad
También declarada tensión inusual o aumento del tono muscular, es considerada
un trastorno motor que refiere a la rigidez o falta de relajo en los músculos
imposibilitando el movimiento parcial o total, a partir de ciertos grupos
musculares que se contraen de forma continua. Los reflejos son más fuertes o
exagerados, tienen movimientos espasmódicos repetitivos (clonos) o tijereteo
(cruce de piernas como si se cerraran las puntas de unas tijeras) interfiriendo
en acciones como caminar, comer o hablar. Esto se aprecia en la postura
anormal, en los ángulos infrecuentes o indebidos de los hombros, brazos,
muñecas y dedos de las manos debido a la rigidez muscular.
Las áreas corporales que más afectan esta patología son:
- En las extremidades inferiores (piernas): afecta fundamentalmente los cuádriceps, gemelos y aductores de la cadera.
- En las extremidades superiores (brazos) afecta fundamentalmente a los músculos flexores de los dedos, muñeca, bíceps y aductores del hombro.
Los individuos que sufren esta patología a menudo la describen como “sensación de pesadez y rigidez en piernas y brazos”.

Imagen
1: Representación de espasticidad en codo, muñeca y dedos.
Fuente: https://www.lifeder.com/espasticidad/

Imagen 2: Representación de espasticidad en codo, muñeca, antebrazo, puño y pulgar.
Fuente: https://www.lifeder.com/espasticidad/
Hiperreflexia:
También llamada disreflexia autónoma (DA) es una reacción anormal y exagerada del sistema nervioso involuntario (autónomo) a estímulos externos o corporales, provocando el aumento alarmante de la presión arterial, la aceleración del número de pulsaciones por minuto, sudoración excesiva, tonos azul grisáceo de la piel, palidez, enrojecimiento, espasmos musculares, entre otros.

Imagen 3: Representación de una respuesta anormal y exagerada del SNI a estímulos externos o corporales (hiperreflexia)
Fuente: http://download.com.d0t.ru//?q
Reflejo de Babinski
Un reflejo es la respuesta del cuerpo a cierto estimulo, en este caso alude a un reflejo normal propio de los neonatos, comienza a desaparecer a partir de los 12 meses y hasta los 2 años de edad, y se presenta una vez frotado firmemente la planta del pie haciendo que el dedo gordo se mueva hacia arriba o hacia la superficie superior del pie y los demás dedos se abren en forma de abanico.
Cuando este reflejo aparece habiendo superado los 2 años sugiere un trastorno del sistema nervioso propio de ELA.

Imagen
4: Representación de la respuesta negativa y positiva a la respuesta del
estimulo conocido como reflejo de Babinski.
Fuente: https://cienciaexplicada.com/reflejos-superficiales-cutaneos-y-mucosos.html
Síntomas a partir de la afección de la motoneurona inferior.
Ø Miastenia:
Palabra que proviene de -mi- del griego μυο que significa “musculo” y ἀσθένεια (asthéneia) que significa “debilidad” su conjunción traduce debilidad muscular, es decir, la reducción de fuerza de los músculos o la manifestación de un rápido cansancio muscular poco común.

Imagen 5: Representación de párpado caído causado por la debilidad muscular.
Fuente: https://unidadaferesis.wordpress.com/tag/miastenia-gravis/
Ø Atrofia muscular:
Es la disminución de tamaño de un músculo hasta su deterioro total. Los signos que indican la presencia de esta característica son, por ejemplo, el que se visualice acentuadamente uno de los brazos o piernas más pequeño que el otro, o poseer una considerable debilidad en una extremidad.

Imagen 6: Representación de la diferencia entre un músculo sano y uno que presenta atrofia.
Fuente: http://slideplayer.es/slide/8648856/
Ø Fasciculaciones:
Cuando debajo de la piel vemos pequeñas contracciones que no producen el movimiento de los músculos estamos viendo fasciculaciones, es decir, descargas de forma espontánea en grupos esqueléticos abastecido por una única fibra nerviosa motora. Es lo que sentimos cuando nos “tiembla” el parpado.

Imagen 7: Representación del movimiento involuntario presente en el párpado del ojo.
Fuente: http://healthydefinition.com/jumping-eye-is-not-a-good-sign/
Ø Hiporreflexia
Al explorar los reflejos bicipitales (percusión sobre el tendón del bíceps con el codo flexionado), triciptales (percusión del tríceps con el codo flexionado), estilo radial (percusión sobre apófisis estiloides radial), rotuliano (percusión sobre el tendón rotuliano) y aquileo (percusión sobre el tendón de Aquiles), se emiten respuestas disminuidas.

Imagen
8: Representación de una respuesta débil frente al estimulo ocasionado a partir
de la percusión sobre el tendón rotuliano.
Fuente: https://www.slideshare.net/g3n3xiitap/motilidad-cintica
Ø Hipotonía o flacidez
El tono muscular se encuentra disminuido revelando excesiva elasticidad, con falta de firmeza provocando que las articulaciones se muestren flácidas aún con el musculo contraído y al moverlo se denota blando y un movimiento muy dilatado.
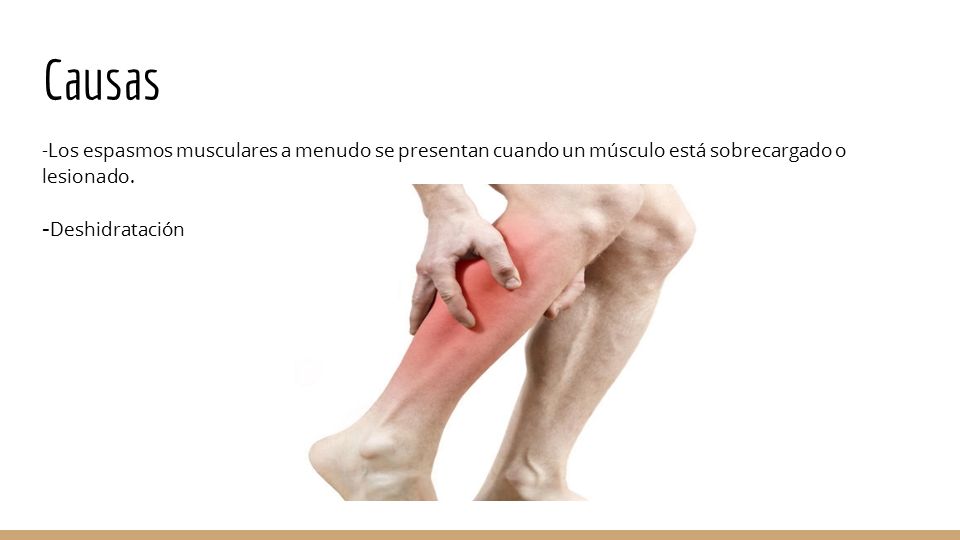
Ø Calambres
Son contracciones o espasmos repentinos involuntarios en uno o más músculos que pueden durar desde unos segundos hasta varios minutos, en algunas ocasiones provocando un gran dolor que se puede aliviar mediante el estiramiento o un suave masaje.

Imagen 9: Representación de una de las zonas que pueden ser afectadas por un calambre.
Fuente: http://images.slideplayer.es/37/10693179/slides/slide_3.jpg
{kind=link}
Síntomas a partir de la afección de la parálisis bulbar progresiva
Ø Disartria:
A partir de una parálisis, debilidad o incoordinación de los órganos de fonación (cavidades glóticas: laringe, cuerdas vocales y resonadores -nasal, bucal y faríngeo) se produce dificultad para hablar. Existen distintos grados severidad de disartria:
Leve – donde el habla es en general claro, se logra comprender lo que el individuo intenta comunicar, pero esto presenta algunas cualidades: problemas en los fonemas, es decir, la articulación de algunos pocos sonidos de su lengua, especialmente en palabras de mayor longitud o aquellos que requieren una coordinación muscular más elaborada. El problema radica en que sustituyen un fonema por otro, lo omiten o al articularlo con inexactitud realizan un sonido ajeno al respiratorio lingüístico del comunicante.
Este tipo de disartria también se muestra a través de un aumento de velocidad (taquilalia) y/o intensidad del habla, provocando la omisión de fonemas o silabas que afectan la inteligibilidad. Muchas veces al comprometerse la movilidad del paladar blando la persona tiene una resonancia hipernasalizada.
Moderada – Se perciben los mismos trastornos que en la disartria leve, pero se incluyen y desvirtúan más sonidos del habla empeorando las dificultades. Por momentos se torna inentendible lo que la persona intenta comunicar debido a la articulación desdibujada y dificultosa que presenta.
Severa – En este último tipo de disartria se torna muy difícil comprender lo que la persona está intentando decir ya que la movilidad de los órganos orolinguofaciales no funcionan de manera efectiva, dando como resultado procesos fono-respiratorio articulatorio y de resonancia fallidos.
Ø Disfagia:
Es un trastorno en la deglución, es decir, el paciente necesita más esfuerzo y demora más tiempo para desplazar la saliva, los alimentos o líquidos desde la boca al estómago o, directamente se le imposibilita realizar dicho movimiento, en algunos casos provocando dolor, babeo, tos, asfixia, entre otros. Puede ocurrir que algunas personas puedan adoptar una determinada posición para comer pero muchas otras necesitan sondas para alimentarse y poder así ingerir las suficientes calorías y líquidos para nutrir el cuerpo.

Imagen 10: Representación del esófago produciendo contracciones normales en el primer esquema, y anormales o débiles en el segundo, debido al daño del funcionamiento muscular del mismo.
Fuente: https://www.slideshare.net/FRAN182/disfagia-y-broncoaspiracion
Ø Sialorrea:
También recibe el nombre de ptialismo (por su nombre en griego πτυαλισμός ptyalismós) o salivación y es un trastorno de la producción de saliva por la cual se segrega saliva o flujo salival en exceso.
Ø Signos Pseudobulbar:
También denominado SPB es un conjunto de signos clínicos que incluyen desórdenes de la deglución y de la fonación, una reducción de movilidad de la lengua y risas o llantos esporádicos. Estos últimos sin provocación alguna ni coherencia con la forma en que el paciente se siente, con frecuencia variable, puede ser una vez a la semana o suceder varios episodios en un mismo día, desde pocos segundos hasta descargas emocionales que duran uno o dos minutos, produciéndose cuando están solos o contras personas, y los pacientes no pueden advertir que van a ocurrir, aunque algunos logran aprender a predecir acontecimientos, pensamientos o estímulos que puedan provocarlos.
Síntomas comunes que se desarrollan en todos los tipos de ELA
Ø Fatiga
Refiere a la falta de motivación, falta de energía, agotamiento constante, debilidad pronunciada o cansancio extremo, que puede hacer que las tareas cotidianas se tornen más dificultosas y que no se alivian con descansar de la manera adecuada, tener una nutrición equilibrada o moverse en un ambiente con un bajo nivel de stress.
Ø Astenia
Es un síntoma muy similar a la fatiga ya que involucran el continuo estado de agotamiento físico y mental, pero incluye además alteraciones de la personalidad, ansiedad y no sólo no mejora con el reposo, sino que además puede emporar afectando funciones intelectuales como menor atención o concentración.
Ø Pérdida generalizada de la masa muscular y el peso corporal
Debido a la dificultad para deglutir o para tragar, o debido a que su metabolismo sube y queman más calorías, los pacientes tienden a desnutrirse.
Ø Contracturas y rigidez que interfieren con la coordinación motora
Pierden la capacidad de caminar normalmente y empiezan a tener limitaciones en las actividades de la vida cotidiana como vestirse, lavarse, entre otros.
Ø Compromiso del sistema respiratorio:
En etapas avanzadas, pérdida de la capacidad de respirar con autonomía, por lo que se precisa un respirador artificial.
Ø Labilidad emocional con inapropiados estallidos de risa o llanto:
Este síntoma comienza a obligarlos a privarse de actividades sociales.
Ø Conservación plena de las habilidades intelectuales o sensitivas:
No se pierden las facultades mentales, sensoriales (la vista, el oído, el olfato o el gusto), las funciones musculares automáticas como los aquellos que realizan el corazón, intestinos, entre otros) y la función sexual.
Ø Control de esfínteres y movimiento ocular:
No es dañado los músculos que controlan la micción y defecación y se conservan los movimientos oculares hasta el final.
¿Cómo se diagnostica ELA?
Al ser una enfermedad comparativamente poco frecuente, en algunos casos, con muy leves síntomas iniciales, inclusive hay algunos a los que sus afectados no le prestan una total atención o que pueden haberle atribuido a otras causas diferentes sus síntomas (como algún tropiezo o por ejemplo la fatiga), se posterga de esta manera la visita a un médico y resulta dificultoso que la ELA sea diagnosticada de inmediato. Asimismo, no existe un estudio especifico que pueda diagnosticar la enfermedad, sino que se requieren muchas observaciones mediante análisis o exámenes médicos para la eliminación de otros trastornos potenciales.
Es necesario obtener la mayor certeza posible en la identificación de los pacientes que padecen la enfermedad, en primer lugar para que la correcta individualización posibilite la más adecuada terapia desde el momento del diagnóstico, en segundo lugar, para que las patologías de similar expresión clínica puedan ser reconocidas y tratadas consecuentemente, y por último, es que las drogas de potencial efecto beneficioso puedan ser administradas a enfermos con certeza diagnostica que hubieran querido participar en ensayos terapéuticos.
Exámenes médicos.
Para comenzar el médico neurólogo solicita al paciente una serie de exámenes a los cuales normalmente se realizan de manera externa, pero también puede requerir para alguno de ellos la hospitalización por un tiempo corto.
A partir del examen clínico, el médico especialista en neurología, basándose en su experiencia, establecerá los signos y exámenes adecuados.
§ Laboratorio.
Generalmente, es solicitado un examen de laboratorio donde examinan la creatina quinasa, una enzima que se acrecienta al dejar de funcionar los músculos, pero este puede ser también síntoma de otras condiciones médicas, obligando de esta manera a realizar otras pruebas como el electromiograma o electromiografía (EMG).
§ Electromiograma (EMG)
La EMG es conocido como el examen de la
aguja, ya que verifica la salud de los músculos y terminales nerviosas que los
controlan introduciendo un electrodo de aguja muy delgado a través de la piel
dentro del musculo. El personal de atención medica solicita la contracción del
musculo, por ejemplo, doblar el brazo y el electrodo en la aguja detecta la
actividad eléctrica liberada por los músculos y permite visualizarla en un
osciloscopio (monitor que muestra actividad eléctrica en forma de ondas) y a
partir de amplificadores oír el sonido la actividad. Este examen se realiza en
cualquier parte del cuerpo y detecta los músculos que no presentan una
actividad eléctrica normal, inclusive pueden observarse las anormalidades
cuando el musculo aun no se
encuentra afectado, constituyendo de esta forma una herramienta muy importante
para el diagnostico.

Imagen 11: Representación de los músculos en el brazo
Fuente: http://neurofisiologiagranada.com/emg/emg-quees.htm

Imagen 12: Representación de los músculos en la pierna.
Fuente: http://neurofisiologiagranada.com/emg/emg-quees.htm

Imagen 13: Representación de los músculos en la región cráneo-facial y el cuello.
Fuente: http://neurofisiologiagranada.com/emg/emg-quees.htm
Otros
estudios que se realizan son aquellos de conducción nerviosa en los cuales a
partir de una almohadilla colocada sobre la piel aplican un impulso eléctrico
para lograr medir la velocidad a la cual los nervios transportan señales eléctricas.
 .
.
Imagen 14: Representación del estudio de exploración del nervio facial.
Fuente: http://neurofisiologiagranada.com/emg/emg-quees.htm

Imagen 15: Representación del estudio de exploración del nervio de la mano.
Fuente: http://neurofisiologiagranada.com/emg/emg-quees.htm

Imagen 16: Representación del estudio de exploración del nervio del pie.
Fuente: http://neurofisiologiagranada.com/emg/emg-quees.htm
§ Estimulación Magnética Transcraneal (EMT).
Además, los especialistas suelen solicitar una Estimulación Magnética Transcraneal (EMT), estudio que mide la actividad de las motoneuronas superiores a partir de un método no invasivo de estimulación de la corteza cerebral. Realiza una suave estimulación del tejido nervioso (corteza cerebral, medula espinal, vías motoras centrales y nervios periféricos), indoloro, interfiriendo de forma controlada en la actividad del cerebro.

Imagen 17: Representación de la estimulación generada en el tejido nervioso a través de ondas magnéticas.
Fuente: https://www.neurociencias30dias.org/revista/neuropsicolog%C3%ADa/
§ Resonancia Magnética (IRM).
Médicos neurólogos solicitan Imágenes por Resonancia Magnética (IRM), un examen imagenológico que emplea imanes y ondas de radio para elaborar imágenes del cuerpo. Cada imagen individual del estudio se denomina cortes, pueden ser desde docenas hasta cientos de imágenes que pueden ser almacenadas en una computadora o impresas en una película.
Para realizarlo es necesario que el paciente no cuente con prendas con broches metálicos, ya que esto puede causar imágenes borrosas y puede que al paciente le coloquen pequeños dispositivos llamados espirales, alrededor de la cabeza, brazos o piernas que ayudan a enviar y recibir las ondas de radio y mejoran la calidad de imágenes. En muchos casos los técnicos ofrecen una bata de hospital para proceder y el paciente ingresa en una maquina cilíndrica para tomar las imágenes internas del cuerpo. Si bien este estudio no determina la enfermedad de la neurona motora, ayuda a descartar otras enfermedades, como por ejemplo un tumor.
§ Otros estudios que pueden ser requeridos:
Punción lumbar: Consiste en la introducción de una aguja en el espacio subaracnoideo de la medula espinal entre la tercer y cuarta vertebra de donde se obtiene el líquido cefalorraquídeo
Biopsia muscular: Proceso por el cual se extrae
una parte de tejido para analizarlo macroscópicamente.
Pero estos últimos sólo se realizan en casos que los hallazgos clínicos indican
que podrían ser útiles.
CADITELA
Esto llevo a la Cátedra de Neurología de la Facultad de Medicina de la UBA, a la Sociedad Neurológica Argentina y al Capitulo Argentino de Lucha contra las Enfermedades de la Motoneurona a convocar a personas con experiencia en diferentes aspectos de la patología para alcanzar un consenso (CADITELA) en lo que respecta al diagnóstico y manejo terapéutico de aquellos afectados por la enfermedad.
En primer lugar se solicita un examen clínico donde el médico especialista en
neurología a partir de su experiencia, reconoce signos y determina los exámenes
adecuados para realizar.
En 1990, en España se elaboró una lista de criterios de diagnóstico a los que
llamaron los Criterios del Escorial, y son revisados periódicamente y
actualizados, según las nuevas concepciones que vayan surgiendo. La última
actualización fue en USA en 1998.
Actualmente los criterios en los que se basan los profesionales para realizar el diagnostico de ELA establecen:
Ø Presencia de:
A: 1 A partir de un examen clínico, electrofisiológico o neuropatológico se evidencie de degeneración del tipo de neurona motora inferior (NMI).
A: 2 A partir de un examen clínico se pruebe una degeneración de neurona motora superior (NMS)
A: 3 En una misma regio o en otras, a partir de la historia clínica o evaluación física, se observe una dispersión progresiva de los síntomas o signos.
Ausencia de:
B: 1 1) Demostración electrofisiológica o patológica de otra enfermedad o proceso que pueda explicar los signos de degeneración de neurona motora superior o inferior.
B: 2 Demostración de neuroimagen de otro proceso o enfermedad, que pueda explicar los signos clínicos y electrofisiológicos explicados.
El CADITELA, además, para la aplicación de los criterios diagnósticos para este tipo de esclerosis recomienda:
§ Para requisar la presencia de la debilidad muscular se debe efectuar la prueba de fuerza manual en los cuatro miembros, tronco y cuello, empleándose las escalas de Kendall o la del MRC:
En esta escala se cuantifica la disnea (falta de aire o dificultad para respirar).

Imagen 18: Representación de la escala modificada del MRC.
Fuente: http://www.tratamientoictus.com/2015/03/14/escala-de-disnea-mrc/
§ Entender los exámenes electromiográficos (EMG) como una extensión del examen neuronal. Son relevantes sólo cuando el profesional actuante tiene adecuado entrenamiento en enfermedades neuromusculares y se efectúan con criterio de información y búsqueda examinando áreas clínicamente no involucradas.
§ La biopsia muscular no es necesaria para el diagnostico de ELA, y estará reservada para:
- La búsqueda de signos de lesión de MNI o no evidenciable por otros métodos.
- Cuando existan dudas respecto del extirpe del proceso (neurógeno, miopático, atrofia muscular por desuso)
- La diseminación de los signos y síntomas no siguieran un patrón de vecindad anatómica, y se desarrollaran en forma salpicada, deberán primero plantearse otros diagnósticos.
- Los estudios de imágenes no son imprescindibles para el diagnostico de ELA y deberán reservarse solo para aquellos casos en los que existan sospechas de otro proceso que pusiera explicar signos clínicos y electrofisiológicos observados.
- Ante el diagnostico de ELA definida, ningún hallazgo en exámenes complementarios de laboratorio invalidara el diagnostico. Estos signos deben estar presentes en cuatro regiones:
· Bulbar: Mandíbula, cara, laringe, paladar y lengua.
· Cervical: Cuello, miembros superiores y diafragma.
· Torácica: Dorso y abdomen (hasta el nivel D6).
· Lumbar: Dorso, abdomen (por debajo del nivel D6) y miembros inferiores.
Niveles de certeza
Se establecen 5 niveles de certeza diagnostica de la ELA
§ En primer lugar, tenemos ELA clínicamente definida cuando se constituye una evidencia clínica de lesión en la motoneurona superior e inferior en la región bulbar junto con al menos dos de las otras regiones espinales (bulbo raquídeo, médula cervical, médula dorsal o médula lumbosacra); o se expresa también, cuando el daño se presenta en tres regiones espinales.
§ En segundo lugar, se establece ELA clínicamente probable donde se constituye una evidencia clínica de daño en la motoneurona superior e inferior en al menos dos regiones con signos de motoneurona superior necesariamente mayores a los de la motoneurona inferior.
§ El tercer nivel de certeza diagnostica es ELA clínicamente probable, sustentada por laboratorio, manifestado cuando los signos de daños de la motoneurona superior están presentes en una región y los signos de motoneurona inferior, se hallan presentes en al menos dos miembros habiéndose excluidas otras causas con estudios de laboratorio y neuroimágenes.
§ El siguiente nivel denominado ELA clínicamente posible se define cuando los signos clínicos de daño de la motoneurona superior e inferior co-existen en una sola región; o cuando los signos de la motoneurona inferior son mayores a los de la motoneurona superior.
§ Y por último la ELA clínicamente sospechosa se observa cuando existen sólo signos de daños en la motoneurona inferior o sólo signos de daños en la motoneurona superior.
Si además de las características que se mencionaron para determinar ELA el paciente posee una función respiratoria anormal, disartria, disfagia, función laríngea anormal, pruebas de fuerza anormales, entro otros síntomas que serán evaluados más adelante, y éstos no se le puede adjudicar a otras razón, ello podría sostener el diagnóstico de la enfermedad; pero si por el contrario, la persona comienza a presentar alteraciones sensitivas, anormalidades esfinterianas, alteraciones visuales o parálisis de músculos oculares, entre otros síntomas clínicos, se desalentaría el diagnostico de ELA.
Tratamientos
Actualmente no es posible disponer de un tratamiento especifico que cure la Esclerosis Lateral Amiotrófica, por lo cual para quienes permanecen acompañados de esta dolencia, es necesario contar con un apoyo profesional desde el punto de vista terapéutico que consiste en paliar o mejorar los síntomas a través de fármacos, o ejercicios para rehabilitar las funciones motoras perdidas que en su conveniente indicación pueden encontrar métodos de remplazo para las mismas; como así también contar métodos alternativos o, igualmente oportuno, el apoyo físico, psicológico y moral, (tanto para el paciente como para quienes lo rodean) de una o más personas para el desenvolvimiento en la rutina diaria, especialmente en los momentos en que la enfermedad ya se encuentra muy avanzada.
Medicamentos.
Podemos agrupar los medicamentos utilizados para el tratamiento de la ELA en dos grupos:
ü El primero constituido por drogas específicamente utilizadas para modificar,
en la medida de lo posible, la evolución de la enfermedad:
Modifican la acción del neurotransmisor excitatorio glutamato que, al acumularse en la sinapsis de la neurona motora ejerce efecto toxico sobre ella, el gabapentin lo hace a nivel presináptico, disminuyendo la síntesis del transmisor, mientras que el Riluzole o la Lamotrigina, dificultan su liberación. El memantine, el dextrometorfan y el topiramato, actúan en la post-sinapsis, bloqueando los receptores de glutamato.
·
Riluzole
De las drogas mencionadas anteriormente, sólo el Riluzole demuestra una
utilidad estadísticamente significativa, ya que logró prolongar la vida y/o
aumentar el tiempo trascurrido desde el diagnostico hasta la necesidad de la
realización de una traqueotomía, por esto es la única considerada en el
tratamiento de la ELA. De todos modos, solo se recomienda su utilización
cuando:
v Se trata de pacientes con ELA probable y definida de acuerdo a los criterios El Escorial.
v Se trata de pacientes con no más de 5 años de evolución de la sintomatología.
v Son pacientes con capacidad vital forzada igual o mayor a 60% de la esperada.
v Son pacientes no traqueotomizados.
v Si alguna persona quisiera comenzar con el tratamiento del riluzole estando exento de los parámetros anteriormente descriptos, deberá consultarlos con su médico tratante quien determinara la conveniencia de acuerdo a una valoración costo – beneficio. Esto ocurre cuando por ejemplo un paciente no tiene menos de 5 años de evolución de la enfermedad pero si un buen estado general, u otro ejemplo serian un paciente con deterioro significativo de su función respiratoria pero que mantiene en buena funcionalidad sus extremidades.
Sólo quedara exento del tratamiento en los pacientes cuyo caso resulte desfavorable la relación riesgo – beneficio, lo cual sucede en caso de:
- Presencia de enfermedades concurrentes e incurables.
- Embarazo.
- Enfermedad renal o hepática grave.
- Aquellos pacientes dependientes del respirador o de un tercero para realizar tareas de la vida cotidiana de manera total.
-
En caso de que la mujer quede embarazada o aparece algún efecto adverso ocasionado por la medicación poniendo en riesgo su vida, deberá de inmediato suspender el tratamiento con riluzole.
La eficacia de esta droga aparenta ser mayor en los primeros estadios de la enfermedad y disminuye en los casos más avanzados o terminales. No es de menor importancia destacar que, en ocasiones, su uso involucra la utilización de recursos económicos que podrían ser utilizados para mayor confort del paciente, como, por ejemplo, la adquisición de un sistema de asistencia ventilatoria.

Imagen 19: Representación de la presentación del medicamento.
Fuente: http://slideplayer.com/slide/10926054/
ü El segundo constituido por drogas utilizadas para mejorar algunos signos y síntomas de la enfermedad:
Es posible reducir el malestar que los distintos signos y síntomas producen en el paciente a lo largo de la enfermedad. Se describen a continuación qué fármacos ayudan de acuerdo a la sintomatología que presenta el paciente:
· Debilidad muscular.
El mono hidrato de creatina puede ser de utilidad, así como también los anticolinesterásicos, pero estos últimos en ocasiones pueden aumentar las fasciculaciones. Otras opciones son la amantadina y el modafinil.
· Calambres.
El magnesio, o si este no fuere eficaz, se utiliza la mexiletina o algunos agentes antiepilépticos con capacidad de bloque de los canales de Na+.
· Sialorrea.
En este síntoma puede utilizarse la amitripitilina, la escopolamina o el glicopirrolato. Otra técnica viable es una inyección de toxina botulínica purificada en ambas glándulas parótidas para disminuir la secreción salival en un 30%.
· Espasticidad.
Para tratar este síntoma se pueden utilizar drogas solas o combinadas. El baclofeno es una de ellas, pero se pueden mencionar también la tizanidina o el dantrolene. Si la administración por vía oral no es del todo efectiva, los pacientes pueden también inyectarse toxina botulínica en los músculos más afectados.
· Signos Pseudobulbares.
Pueden tratarse con amitriptilina, foxetina u otros antidepresivos.
· Trastornos de la ansiedad.
En caso de que el paciente requiera un tratamiento ansiolítico se aconseja emplear tranquilizantes menores, como el alprazolam o clordiazepóxido. Pueden usarse también antihistamínicos para evitar el uso de sedantes.
· Dolor:
Síntoma común en los estadios terminales, para el cual deberán suministrarse antiinflamatorios y donde la kinesioterapia juega un papel importante para advertir su aparición.
Ejercicios
Es necesario que los pacientes diagnosticados con ELA realicen ejercicios de movilidad para todas las articulaciones al menos dos veces al día.
Deben mantener erguida su posición, permanecer de pie, y cambiar a menudo de postura para prevenir la rigidez y promover la actividad ósea.
Ayuda el participar de un programa de ejercicios adecuado a las necesidades y capacidades del paciente: nadar, andar, hacer ejercicios sobre bicicleta estática, o en algunos casos, mantener las actividades cotidianas. Es primordial evitar, en la medida de lo posible, la fatiga.
Se pueden establecer algunas instrucciones generales para realizar estos ejercicios:
a. No se debe forzar el ejercicio, sino que deben efectuarse de manera suave y lenta.
b. Puede hacerlos de manera activa, pasiva o con asistencia, de acuerdo a la capacidad de cada paciente.
c. Deben ser repetidos tantas veces le permita el límite de fatiga del paciente
d. No se debe continuar si provocar algún tipo de dolor articular.
Se mencionan a continuación 13 ejercicios para que el paciente con ELA pueda realizar con control y/o intervención profesional, y así prevendrá o al menos retrasará el progreso de la enfermedad.
1. Flexión de cadera y de rodilla
El paciente debe llevar la rodilla hasta el pecho y al mismo tiempo aproximar su talón hacia el glúteo, debe tratar de llevar la punta del pie hacia arriba. Es necesario que la rodilla permanezca derecha evitando que vaya hacia adentro o hacia afuera.

Imagen 20: Representación de la forma de realizar el ejercicio N°1.
Fuente: http://www.araela.org/ejercicios-fisioterapeuticos-para-los-pacientes-con-ela/
2. Giro de la cadera con la cadera doblada
El paciente debe disponer las articulaciones de cadera y rodillas en un ángulo de 90°, y conservando el musculo fijo, mover el pie hacia adentro y hacia afuera deteniéndose en caso de aparición de dolor.

Imagen 21: Representación de la forma de realizar el ejercicio N°2.
Fuente: http://www.araela.org/ejercicios-fisioterapeuticos-para-los-pacientes-con-ela/
3. Cadera y rodilla: flexión y/o extensión; ruedo de la cadera
El paciente deberá flexionar la rodilla para arriba hacia el pecho formando un ángulo de 90° y permaneciendo con la rodilla doblada deberá rotar la pierna hacia él la derecha y luego hacia la izquierda. Es importante respetar el límite de resistencia y detenerse en caso de dolor. Este es un ejercicio que combina los dos mencionados anteriormente.

Imagen 22: Representación de la forma de realizar el ejercicio N°3.
Fuente: http://www.araela.org/ejercicios-fisioterapeuticos-para-los-pacientes-con-ela/
4. Separación de cadera con la rotación neutral
El paciente deberá mantener fija una pierna sobre la camilla, con la rodilla flexionada o no, y la otra pierna con la rodilla tensa deberá llevarla hacia adentro y hacia afuera, sin elevarla mucho, sólo lo justo para que se deslice.

Imagen 23: Representación de la forma de realizar el ejercicio N°4.
Fuente: http://www.araela.org/ejercicios-fisioterapeuticos-para-los-pacientes-con-ela/
5. Flexión dorsal del tobillo con la rodilla recta
El paciente deberá, con la rodilla estirada, disminuir el ángulo entre el pie y la pierna acercando los dedos hacia la parte anterior de la canilla de la pierna (opuesta a la pantorrilla), sin elevar el talón de la camilla. Combinar también este ejercicio con una pequeña fuerza para realizar el movimiento contrario: la plantarflexión, es decir, aumentar el grado entre el pie y la pantorrilla.

Imagen 24: Representación de la forma de realizar el ejercicio N°5.
Fuente: http://www.araela.org/ejercicios-fisioterapeuticos-para-los-pacientes-con-ela/
6. Rotación del tronco
El paciente debe flexionar las rodillas y juntarlas para luego mover las piernas hacia su derecha y su izquierda, sin mover los pies y el tronco.

Imagen 25: Representación de la forma de realizar el ejercicio N°6.
Fuente: http://www.araela.org/ejercicios-fisioterapeuticos-para-los-pacientes-con-ela/
7. Extensión del codo
El paciente deberá flexionar y estirar el codo, preferentemente permanecer con la mano abierta al extender y cerrada al doblar el codo.

Imagen 26: Representación de la forma de realizar el ejercicio N°7.
Fuente: http://www.araela.org/ejercicios-fisioterapeuticos-para-los-pacientes-con-ela/
8. Flexión del hombro
El paciente deberá con el codo extendido, elevar el brazo completo por encima de la cabeza.

Imagen 27: Representación de la forma de realizar el ejercicio N°8.
Fuente: http://www.araela.org/ejercicios-fisioterapeuticos-para-los-pacientes-con-ela/
9. Rotación interna y externa del hombro
El paciente deberá apoyar el brazo sobre la cama, con el codo flexionado, llevará la mano y el antebrazo hacia atrás y hacia adelante.

Imagen 28: Representación de la forma de realizar el ejercicio N°9.
Fuente: http://www.araela.org/ejercicios-fisioterapeuticos-para-los-pacientes-con-ela/
10. Flexión y extensión de los dedos
El paciente deberá estirar y doblar todos los dedos mientras abre y cierra su mano.

Imagen 29: Representación de la forma de realizar el ejercicio N°10.
Fuente: http://www.araela.org/ejercicios-fisioterapeuticos-para-los-pacientes-con-ela/
11. Movimientos de pie y tobillo
El asistente deberá ahuecar el talón del pie en la palma de su mano y presionar su antebrazo contra el fondo de los pies, tirando el talón hacia arriba.

Imagen 30: Representación de la forma de realizar el ejercicio N°11.
Fuente: http://www.araela.org/ejercicios-fisioterapeuticos-para-los-pacientes-con-ela/
12. Rotación lumbar
El paciente deberá dejar la pierna que se estirará con la rodilla doblada, permitiendo que el pie se apoye a la altura de la rodilla contraria, pero cruzándola por encima de ella. El asistente efectuará fuerza con una mano sobre la rodilla de la pierna que se estirará hacia el lado contrario, y al mismo tiempo con la otra mano sujetará la pelvis (cirulo rojo) impidiendo la inclinación al realizar el movimiento. El paciente percibirá una sensación de tirantez o estiramiento en la parte externa de la pierna que esta cruzada por encima con la rodilla flexionada. Si comienza a sentir dolor en la zona de la ingle, deberá realizarse el ejercicio con menor intensidad.

Imagen 31: Representación de la forma de realizar el ejercicio N°12.
Fuente: http://www.araela.org/ejercicios-fisioterapeuticos-para-los-pacientes-con-ela/
13. Estiramiento de tendón de la corva
El paciente deberá mantener la rodilla estirada, levantar toda la pierna recta junto con el pie también hacia arriba.

Imagen 32: Representación de la forma de realizar el ejercicio N°13.
Fuente: http://www.araela.org/ejercicios-fisioterapeuticos-para-los-pacientes-con-ela/
Debilidad del manejo del musculo del cuello
Al tener debilidad en el cuello se les genera a los pacientes una movilidad limitada de la cabeza, dado que la misma pesa alrededor de 9 Kg. Al poseer debilidad en el musculo del cuello, el paciente es vulnerable a la lesión ante cualquier estiramiento repentino hacia adelante, hacia atrás o de lado, pudiendo causar también una lesión seria en la medula espinal o a las vértebras cervicales de esa zona. Estos movimientos repentinos pueden ocurrir al levantarse o acostarse sobre una cama, o al pararse o sentarse en una silla, causando un gran dolor y en caso de lesión puede también, en algunas ocasiones, afectar la respiración, deglución y la comunicación.
Estas complicaciones y lesiones pueden ser prevenidas o tratadas con estrategias para mejorar la postura y promover su movilidad:
En primer lugar, es esencial mantener una buena alineación del cuerpo al sentarse lo cual se puede ayudar colocando una almohadilla o una toalla rodada a modo de amortiguador, o detrás del cuello para mejorar la postura. También pueden colocarse almohadillas sobre cada apoyabrazos lo cual le proporcionará una máxima comodidad y posición erguida si necesita estar sentado durante mucho tiempo, esto ayuda además a que los pulmones se amplíen proporcionando una mejor respiración.
El paciente puede abastecerse de un collar cervical que lo ayudará a sostener la cabeza cuando se transfiere de un asiento a otro, cuando camina o circula en coche, evitando un movimiento repentino y al permitir una mejor línea visual puede también disminuir el riesgo de caer.
Hay distintos tipos de cuellos cervicales, por lo cual el paciente deberá identificar cual es el apropiado, siendo el más común un collar suave de espuma. Generalmente, si es solicitado bajo orden médica el cuello cervical tiene el costo del mismo cubierto por el seguro médico, pero en caso de que esto no ocurra, pueden obtenerse en la mayoría de los centros de ortopedia.
En estadios más avanzados de la enfermedad donde la persona se encuentra en una silla de ruedas para mantener el cuello firme, a pesar de la debilidad y/o malestar que presente puede apoyar la cabeza sobre la almohadilla presente en la parte de atrás, utilizando una venda como soporte a través de la frente.
Al momento de dormir, es importante contar con una almohadilla que no sea demasiado alta e incluso es preferente la utilización de una toalla rodada por debajo de la parte posterior del cuello, con la cabeza reclinándose levemente sobre una almohadilla baja.
Si el paciente persiste con dolor o dificultad para mover su cabeza y cuello puede consultar con su médico para la prescripción de una terapia física. Algunos tratamientos pueden incluir el uso del calor, masajes, entre otros.
Alimentación
Si
bien un paciente que padece ELA no cuenta con una dieta específica para el
tratamiento de su enfermedad, es primordial que mantenga una alimentación
balanceada, realizando tres comidas diarias, con una participación equilibrada
de los principales grupos de alimentos y que incluyan alimentos ricos en
fibras.
Debe beber al menos 6 a 8 grandes vasos de líquido cada día, evitando bebidas
con cafeína debido a su efecto diurético.
Debe controlar periódicamente el peso con la finalidad de verificar que el aporte de las calorías sea el suficiente para evitar la pérdida del buen estado de nutrición.
Se deben incluir sólidos, semisólidos y/o líquidos (evitando los derivados lácteos), dependiendo la capacidad de deglución del paciente. Si el paciente no logra deglutir los alimentos será obligatorio recurrir a la alimentación por gastrostomía.
Asistencia respiratoria.
En etapas avanzadas de la ELA, el paciente necesitará de asistencia artificial para poder respirar.
I. Oxigenoterapia.
Existen terapias, como la del uso de O2 llamada oxigenoterapia, utilizadas sólo en algunos de los pacientes que presentan debilidad muscular respiratoria e hipo ventilación. Esto va a depender específicamente de cada caso y sólo se administrará bajo la indicación controlada del médico tratante.
II. Ventilación no invasiva (VNI)
Posibilita regular de manera independiente los niveles de presión inspiratoria y espiratoria (BiPAP).
Generalmente es indicada en pacientes consientes, padeciendo una ELA que progresa lentamente y conserva la capacidad de comunicarse y desarrollar algunas actividades cotidianas, pero la capacidad vital forzada se ubica debajo del 50% del valor normal señalando la presencia de insuficiencia respiratoria sintomática. El fin de estos equipos es aliviar los síntomas, normalizar el sueño, mantener la saturación arterial de O2, entre otros, disminuyendo el número de internaciones o el tiempo de ellas, aumentando la calidad y expectativa de vida.
III. Traqueotomía
Es una práctica que constituye la ventilación invasiva, dado que consiste en realizar una incisión en la tráquea para formar una abertura donde se colocara un tubo Será indicada cuando:
La ventilación a partir del BiPAP no es posible o fracasa.
- Cuando el paciente prefiere esta cirugía.
- Cuando se desea una sobrevida mayor.
Steven Hawking: Ejemplo de lucha y esperanza.
Nacido el 8 de enero de 1942 en Londres, fue el mayor de cuatro hermanos, hijo de un padre biólogo investigador y una madre secretaria en investigación médica, lo cual probablemente sea el principio de su interés en la ciencia.
El acreditado físico teórico, astrofísico, cosmólogo, divulgador científico británico y autor de la Teoría del Bing Bang, entre otras obras, nos sorprende por su asombroso intelecto y es, en la actualidad, una de las personas que padecen Esclerosis Lateral Amiotrófica.
En
1962, al poco llegar a Cambridge, comenzaron a aparecer los primeros síntomas y
en el siguiente año, durante su estadía en Oxford (con 21 años) fue cuando le
diagnosticaron ELA, dándole un pronóstico de vida no mayor a 3 años (pronostico
normal de la enfermedad), noticia que, por temor al tiempo anunciado, aceleró
su primer matrimonio. De igual forma esta enfermedad no le impidió mantener su
actividad científica y publica. Al ir perdiendo el uso de sus extremidades
desarrollo en su mente un sistema de resolución de ecuaciones sin la necesidad
de escribirlas.
En 1985 mientras estaba en Ginebra- Suiza, fue internado por una neumonía que
ponía en riesgo su vida, donde los médicos del hospital le realizaron una
operación (traqueotomía) que le restringiría el habla. Desde entonces utiliza
un sintetizador de voz para comunicarse (un productor artificial del habla).
Este dispositivo lo acciona a partir de una contracción voluntaria de una de
sus mejillas componiendo frases y palabras. Poco a poco fue disminuyendo la
capacidad de movimiento en sus extremidades, así como el resto de su
musculatura voluntaria reduciendo al extremo la fuerza y movilidad de su cuello
lo cual le imposibilita mantener su cabeza erguida dejándolo con un movimiento
casi nulo. Hoy efectúa leves movimientos de cabeza y ojos con los cuales
controla un ordenador que maneja su silla de ruedas.
Por motivos desconocidos, a pesar de que el progreso de la enfermedad no se detuvo, hoy 54 años posteriores al diagnóstico, Stephen gracias a los cuidados a los que ha sido sometido a lo largo del tiempo y sus pensamientos positivos acompañados de gran optimismo, permiten que siga asombrando al mundo día a día.

Imagen 33: Fotografía de Stephen Hawking en el año 1980.
Fuente: http://www.hawking.org.uk/images.html
Marco teórico III
Epidemiologia
La esclerosis lateral amiotrófica es una enfermedad que lleva a la muerte en el 50% de los casos en los primeros 3 años posteriores al diagnóstico, siendo ésta la condición más frecuente en adultos, pero no al no ser considerada una enfermedad de declaración obligatoria a nivel nacional, surge la necesidad de implementar un sistema de vigilancia para así conocer su comportamiento en el país.
Bajo encuestas realizadas se pudo determinar la falta de información existente a nivel poblacional, dado que a pesar de la enorme campaña “Ice Bucket Challenge” es muy poca la gente que conoce acerca de esta patología.
Durante el mes de octubre y noviembre de 2017 se llevó a cabo la recolección de datos a partir de encuestas realizadas en CABA y Gran Buenos Aires. Se encuestó a un total de 100 personas al azar, de diferentes edades de las cuales 36 eran hombres y 64 mujeres.

En el grafico se representa el porcentaje de hombres encuestados (36%) y de mujeres encuestadas (64%)
Según los datos obtenidos se puede observar:
Tabla 1
Tabla de Encuestados
|
|
Saben acerca de la enfermedad |
No saben acerca de la enfermedad |
Total |
|
Hombres |
7 |
29 |
36 |
|
Mujeres |
21 |
43 |
64 |
|
Total |
28 |
72 |
100 |
Datos obtenidos en el campo (Elaboración propia)
Se observan porcentajes a partir del motivo por el cual los individuos interrogados conocen acerca de ELA.

En el grafico se puede observar que la mayor parte (75%), no conoce acerca de la enfermedad mientras que los que sí la conocen son un porcentaje mucho menor divididos en aquellos que lo conocen por la publicidad (9%), aquellos que la conocen porque sus estudios se relacionan en algún grado con la medicina (8%) y, por último, aquellos que poseen un familiar o conocido que la padece.
ReNELA
Como se mencionó anteriormente es de suma necesidad implementar un sistema de vigilancia para informar y así obtener registros epidemiológicos. Para esto se realizó una prueba piloto del Registro Nacional de Esclerosis Lateral Amiotrófica (ReNELA) entre junio de 2015 y mayo de 2016, donde se logró crear una base de 215 personas que padecen la enfermedad. La misma se obtuvo a partir del aporte de una red de 35 neurólogos ubicados en diversas provincias del territorio nacional que incluyó historias clínicas de pacientes diagnosticados. Debido a que no se puede tomar como una muestra representativa del país no se pueden establecer estadísticas generalizables a toda la población. Sin embargo, la realización de este proyecto orientador permitió detectar las enormes dificultades que implica la construcción de un registro, dada la heterogeneidad de factores por los que se encuentra influida su implementación y los diversos actores involucrados.
Según el género se distribuyen en 54% hombres y 46% mujeres que se presume que padecen ELA en el territorio Argentino.


En este grafico se visualiza la distribución de pacientes diagnosticados con ELA según provincia de residencia. República Argentina 2015 – 2016
Incidencia y Prevalencia
Esta prueba piloto arrojo además una potencial incidencia de 2 por cada 100.000 habitantes de la población general mientras que se presume una prevalencia de 7 por cada 100.000 habitantes.
Se cree, además que la ELA afecta a adultos de cualquier edad, siendo mayores de 40 años el grupo de los diagnosticados, más frecuentemente entre los 50 y 70 años. De ellos 2 hombres por cada 1 mujer se ve afectados, pero esto se iguala a partir de los 70 años.
ELA en España
En España, sí se logró realizar estudios epidemiológicos que confirman datos donde se visualiza que la ELA es más frecuente en hombres (3 hombres por cada 2 mujeres) aunque a partir de los 60 años estas diferencias se emparejan.
El inicio de la enfermedad es a partir de los 55 años, siendo el 80% de los casos manifestada entre los 40 y 70 años sin importar razas o etnias.
En el reino de España, la incidencia, es decir, el número de casos nuevos en un periodo de tiempo determinado, se presume dos casos por cada 100.000 habitantes al año.
Y si hablamos de prevalencia, es decir, el número de personas afectadas en un período de tiempo es de 1 por cada100.000 habitantes, lo cual significa que unos 40.000 españoles vivos desarrollarán la enfermedad.
Estas son estadísticas que en nuestro país por la falta de datos (directamente vinculado a la falta de información existente) no se pueden establecer con certeza ya que no se podría tomar una muestra significativa si consideramos a la ELA como una enfermedad rara en vez de una enfermedad de carácter de notificación nacional obligatorio (ENO). A partir del proyecto efectuado por la Asociación de Esclerosis Lateral Amiotrófica llamado ReNELA se impulsa a poder recolectar los datos y los recursos necesarios para combatir y dominar a la enfermedad.
¿Cómo inscribirse al ReNELA?

Imagen 34: Representación explicativa de los pasos a seguir para formar parte del ReNELA.
Fuente: http://www.asociacionela.org.ar/index.php/la-ela/registro-nacional-de-ela
Conclusión
A partir de los datos recolectados y la información observada desde las diferentes investigaciones realizadas, podría decirse que es una enfermedad a la que se le debe brindar mayor atención de la que recibe, ya que a pesar de que se logró encontrar una variedad de medicamentos que ayudan a mejorar la calidad de vida, o tratamientos y ejercicios con estos mismos fines, continua presente la necesidad de una difusión que pueda concientizar a los individuos.
Si
bien la ELA no tiene una cura establecida, para que esto pueda ser lograrse en
un futuro próximo, es sumamente necesario poder atribuir mayores presupuestos y
mejores medios para la investigación de esta enfermedad en nuestro país, evitando
de esta manera que los médicos tengan que decidir sobre los procedimientos más
convenientes a seguir, comparando beneficios y riesgos de cada una de las
opciones.
Impulsando a la propagación de información, tanto a la sociedad como a la
comunidad científica, podrían lograrse mayores objetivos.
Agradecimientos
Agradezco a mi familia y mis amigos que fueron mi sostén en cada día, me alientan en todo momento y creen en mí de manera incondicional.
Y agradezco particularmente a la Prof., Adriana Gargaglione, por la ayuda concedida.
Dedicatorias
Este trabajo se lo dedico a mi ahijado, a quien considero una de las personas más importantes de mi vida, a mis compañeras de instituto de quienes, además, me llevo una amistad y, por último, pero muy significativo: a mi mamá, a quien extraño siempre.
Bibliografía
Sitios Web:
-https://kidshealth.org/es/kids/als-esp.html
-http://adelaweb.org/la-ela/la-enfermedad
-https://es.slideshare.net/verorosso/transmisin-del-impulso-nervioso-sinapsis
-http://naukas.com/2015/04/20/las-neuronas-del-movimiento/
-http://biologiablog-biologicblog.blogspot.com.ar/2010/12/conceptos-tipos-y-definiciones-de-ela.html
-http://www.asociacionela.org.ar/images/stories/PDF/InformefinalconsolidadoReNELA.pdf
-https://medlineplus.gov/spanish/ency/article/003297.htm
-https://www.lifeder.com/espasticidad/
-https://medlineplus.gov/spanish/ency/article/001431.htm
-https://es.healthline.com/health/atrofia-muscular
-https://elmundodelmusculo.blogspot.com.ar/2012/12/dolencias-y-enfermedadesmusculares-un.html
-https://medlineplus.gov/spanish/ency/article/003296.htm
-https://neuropeditra.org/2016/11/07/que-es-el-tono-muscular-hipotonia-e-hipertonia
-https://www.ineco.org.ar/disartria/
-https://medlineplus.gov/spanish/ency/article/003088.htm
-http://sid.usal.es/idocs/F8/FDO7213/ELA.pdf
-http://www.asociacionela.org.ar/
-http://www.hawking.org.uk/images.html
-http://neurofisiologiagranada.com/emg/emg-quees.htm
Libros:
El Tratado De Enfermería MOSBY – BEARE / MYERS Vol. 3 Editorial Oriente s.a.
AUTORA:
TSAC, (Estudiante) María Victoria Guntren
COAUTORA
Prof. Silvina Pérez, Especialista en sobrepeso y obesidad
Coach en Nutrición y Educación
INSTITUTO:
Instituto de Formación Técnica Superior N. º 10, CABA
RECURSO HUMANOS:
TSAC, (Estudiante) María Victoria Guntren
ESTRATEGIAS:
Recopilación de datos a partir del acceso a libros y varios sitios web.
COSTOS:
Tiempo: Aproximadamente 2 Meses.
Gastos: Mínimos
| Este apunte fue enviado por su autor en formato DOCX.
Para poder visualizarlo correctamente (con imágenes, tablas, etc) haga click aquí o aquí si desea abrirla en ventana nueva. |
Boletín de Novedades

Acceso rápido
• Monografias
• Examenes
• Enlaces
• Publicar material o sitio
• Foros
• Diversion
• Test de Orientacion
• Institutos
• Secundarios
• Universidades de Argentina
• Residencias Universitarias
• Guia de Carreras Universitarias y Cursos
• Cultura del Mundo
• Buscador en tu sitio
 Tene tu sitio web facil de usar. Aparece en Google! Tene tu sitio web facil de usar. Aparece en Google! |
| LA GUERRILLA DIGITAL |
| Intercambios Virtuales |
Intercambio de enlaces
Sobre ALIPSO.COM
Monografias, Exámenes, Universidades, Terciarios, Carreras, Cursos, Donde Estudiar, Que Estudiar y más: Desde 1999 brindamos a los estudiantes y docentes un lugar para publicar contenido educativo y nutrirse del conocimiento.
Contacto »Legales